![]() Biology
C2005 Lecture 6
Biology
C2005 Lecture 6
::Protein purification methods::
PROTEIN PURIFICATION/SEPARATION METHODS
::ultracentrifugation::
Here is one sometimes useful method: ULTRACENTRIFUGATION
Ultra means = >20,000 rpm; 60,000 rpm is common, compare a Ferrari revving
at 6000 rpm, red-lining; this is ten times faster; you need a vacuum chamber
so no heat from air friction will be produced.
Diagram of tube, spin, distribution of molecules ... ...
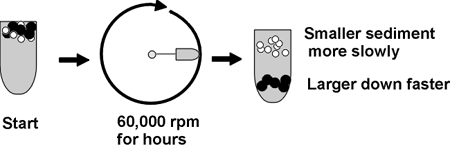
A mixture of molecules will be subject to two main forces in the ultracentifuge
as it starts to spin (ignoring buoyant force):
Causing sedimentation
is the centrifugal force = m(omega)2r
= (which is proportional to the mass or MW of a protein).
m = mass, omega = angular velocity, and r = distance from
the center of rotation.
Opposing sedimentation
= friction = foV.
fo = frictional coefficient, a constant for any
particular protein, it is minimum for a sphere, higher for less compact
shapes like cigars or pancakes.
V = velocity of the molecule as it moves away from the center
of rotation.
Soon after accelerating, V increases to a point where
no further acceleration takes place, as the forces on the molecule are
balanced. It continues to sediment, but at
a constant velocity.
Now at this point, at this velocity: Centrifugal Force = Frictional force
(there's no net force, no acceleration, but
constant velocity)
So at this point (soon achieved): M(omega)2r = foV
And: V = m(omega)2r/fo,
where f = a frictional coefficient dependent on shape (to visualize the
effect of shape on friction, compare the velocity of a falling feather
vs. a tiny pebble of equal weight, dropped in the fluid of air).
Higher f = more friction.
If we assume a spherical shape, then we can estimate a MW (Assume fo,
and then measure V and r, so we can solve for m, or the MW)
On the other hand, if we know the MW, we can get information about shape
(via fo).
Sedimentation velocity is often measured in Svedbergs, which takes the
centrifugation conditions into account s = V/(omega)2r, and
so m = sfo.
So ultracentrifugation separates proteins on the basis of MW and shape.
It is a gentle procedure (non-denaturing, can be carried out at nice low
temperature (say 4 deg C, which tends to stabilize proteins) and in the
presence of a buffer at pH 7 and physiological levels of salts).
You can recover your protein by punching a hole in
the bottom of the centrifuge tube, and collecting the solution in a series
of tubes as it drips out the bottom. Each tube can then be examined, or
assayed, for the presence of the protein to be purified. For this purpose
you need to be able to detect the protein in the midst of the other proteins.
For example, if you were purifying Anfinsen's ribonuclease, you could
measure the ability of the tube contents to catalyze the breakdown of
RNA to its monomers.
How about separation on the basis of the net charge of a protein. We separated
amino acids on the basis of charge in paper electrophoresis. For proteins,
the solid supporting material is a gel, not paper:
GEL ELECTROPHORESIS:
There are two types -
::native gel electrophoresis::
First: native gel electrophoresis
acrylamide, a reactive chemical (a monomer in this chemistry) in aqueous
solution can be made to polymerize into polyacrylamide (thus polyacrylamide
gel electrophoresis, or P.A.G.E.). The result
is a network of polymer fibers, which form a gel, with about the consistency
of Jello.
Usually carried out in an apparatus in which this gel stands vertically
supported in a sandwich between two glass plates. The top of the gel is
connected by a salt solution to an electrode, as is the bottom edge of
the gel. A protein mixture is applied to the top of the gel slab.
The electrophoresis is started by applying a voltage (~200 v) across the
gel.
The rate of migration of a protein in the gel depends on two properties:
The first is its net charge.
Molecules with the most charge (of a sign opposite to that of the far
electrode) will migrate to the far electrode fastest.
Second, its "size" (which is proportional to MW
if spherical). The gel consists of a network of fibers. Depending on the
concentration of polyacrylamide, the network can be dense or tight, so
that the proteins have trouble migrating, as they must negotiate their
way through the tangled fibers. Molecules that are smallest (i.e., lowest
MW) will worm their way through the gel fibers fastest.
So the smallest and most highly charged protein wins the race.
After the electrophoresis has been stopped, molecules will be distributed
along the gel length according to these two characteristics (MW and net
charge). For instance, a highly charged protein molecule, although pulled
with a greater electromotive force, will not have gotten very far if it
is relatively large.
[Note that molecules with a charge opposite to the near electrode, will
migrate up and off the gel, into the buffer reservoir and be lost. Trial
and error will dictate how you setup the electrophoresis if you do not
know the charge on the protein you are trying to isolate.]
Sometimes the gel is made purposely loose, so that the effective pore
size is very large and then charge alone determines mobility. Starch gels
rather than polyacrylamide have been used to create large pore gels.
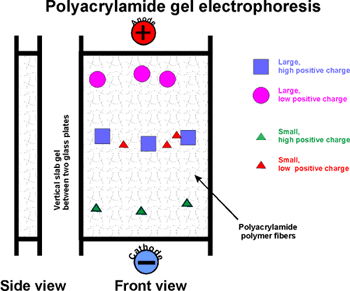
::SDS gel electrophoresis::
A second, more widely used variation of gel electrophoresis is SDS
PAGE.
Here sodium dodecyl sulfate, or SDS (or SLS) is included
in the gel: CH3-(CH2)11-SO4=
[sulfate is similar in structure to phosphate, and is a strong acid].
Like a phospholipid, SDS has a highly polar end and a highly hydrophobic
body.
Might you expect SDS to denature a protein? Yes. It's a detergent and
a powerful denaturant. It binds all over the protein, coating every protein
with a nearly uniform negative charge. SDS
is put into in the gel when you form it and into the electrophoresis buffer.
Now when SDS-PAGE is run, where should the anode be placed? Does it matter?
Yes, the protein is coated with negative charge now, so the anode is always
the far electrode, put at the bottom.
Under these denaturing conditions, the polypeptides exist as a random
coils, which then migrate solely on the basis of their size, which is
the equivalent of a sphere for all polypeptides. Larger molecules have
more difficulty finding their way through the polyacrylamide fibers. So
the lowest MW wins and always wins in SDS-PAGE.
If you run standards of known MW, you can determine the MW of your protein
by comparison, and this is a very common way to assign a MW to a polypeptide.
However, it is not always completely accurate, as some proteins probably
do bind a bit more SDS than others. And one must remember to reduce the
disulfides with mercaptoethanol first (usually), so as to have a truly
random coil for comparisons with other proteins.
If you don't yet know what a protein does, you can just call it by its
molecular weight, from SDS gels: e.g., p53, a famous protein whose absence
is associated with cancer was named this way, and the name has stuck even
though quite a lot is known about its function now (p in p53 stands for
protein, so you have names like p27, p100 etc.).
::Gel filtration::
If you have a protein with quaternary structure, SDS-PAGE will give you
the MW of the polypeptide subunits, since the SDS will denature the protein
and so dissociate it into its subunits. If you want to know the MW of
a protein in its native quaternary structure, you need a method seprates
proteins under mild conditions that maintain its native structure.
For this we could use molecular sieve chromatography,
also called or Sephadex chromatography, or
gel filtration (these are all ~synonymous).
You start with plastic-like beads in a glass column with a support screen
on the bottom.
You add your protein mixture to the top of the column material, then begin
elution by adding a large volume of a buffer. The beads have been manufactured
to be riddled with channels of a specified fairly uniform size. If a protein
is smaller than the channel size, it enters a channel, explores it by
diffusion, and eventually makes its way back out, having wasted its time
in the race to the bottom of the column. Larger proteins can't fit in
to the channels, so they don't waste their time, and they win the race.
Intermediate sized proteins have a bit of trouble getting into a channel,
so they waste some time but less than the smaller proteins. So larger
molecules come out (elute) first, and the smallest come out last. Here
again, you would collect the eluted proteins in a series of tubes, and
then assay each tube for the presence of the protein being purified. If
you calibrate the column by noting the behavior of spherical proteins
of known size, you can determine the MW of your protein by comparison,
if it is also spherical. If is is not spherical it will appear to have
a higher molecular weight than its true MW (imagine a pancake being excluded
from a channel while a sphere of the same MW gets in).
Other methods include ion exchange chromatography,
which also takes advantage if the net charge on a protein, and affinity
chromatography, which takes advantage of the surface properties of a protein
(which we'll discuss next). One can purify a particular protein away from
all other proteins in 4-5 such steps. For more on these protein separation
techniques, see the protein
separation handout.
Here are some images of laboratory ultracentrifuge,
slab gel electrophoresis,
gel filtration apparatuses.
One has to able to follow the protein of interest, to detect its presence
in the presence of all others. One often uses its functional properties
for this, which brings us back to structure and function
of proteins.
::Membrane proteins::
As an introduction into an example of the function of proteins, let's
consider first a special class of proteins that do not follow some of
the rules we have have seen that govern protein structure in general.
These are the MEMBRANE PROTEINS, the proteins
that reside, not in the aqueous environment of the cytoplasm or the nucleus,
but rather in the hydrophobic environment of the membranes of the cell,
including the cell membrane.
In these proteins, the hydrophobic side chains are on the outside, as
there are no hydrophobic forces present i to force then to coalesce. These
proteins can usually diffuse laterally in the lipid bilayer of the membrane
, they can aggregate with each other with specificity, and they can become
anchored via attachment to structural cytoplasmic fibers. They can be
nearly completely enveloped by the lipid bilayer, or they could be partially
immersed, with a more conventional half (i.e., hydrophobics on the inside,
hydrophilics on the outside) sticking out into the cytoplasm or on the
outside of the cell. See [Purves6ed
5.1], [Purves6ed
5.4].
One class of membrane proteins act as channels through the membrane. The
channel proteins are formed into cylinders, with a hydrophobic exterior,
but with hydrophilic groups lining the hole through the interior of the
cylinder. Small molecules can pass through the cylinder, or channel, but
larger molecules cannot fit through (note: macromolecules CANNOT diffuse
onto cells). See [Purves6ed
5.9]
So we have a cellular function for a protein, and a function that shows
some selectivity on the basis of size there. How about other criteria
of selectivity, like charge?
Some channels can distinguish charge: + repels +, and attracts -, so a
channel that is lined with positive charge could bind a negatively charged
ion and, if there's a higher concentration outside than inside, eventually
pass these ions along from the outside of the cell to the inside. A positively
charged ion on the other hand would be repelled by the channel and so
would not get into the cell by this route.
You can a imagine the same sort of selectivity based on hydrogen bonding,
for example.
So a protein can detect charge and surface electrical properties, but
how about shape?
::Proteins bind molecules
with great specificity::
Consider a pocket on the surface of a folded protein. As I've drawn this
surface pocket, the free amino acid gly can fit in and bind using electrical
attraction or ionic bonds. However, the closely related amino acid alanine,
with a methyl group as a side chain, cannot fit into this hypothetical
protein A drawn here (yellow). Ionic bonds and van der Waals bonds (VDW)
are responsible for the binding in this example so far (using different
colors for protein and aa)
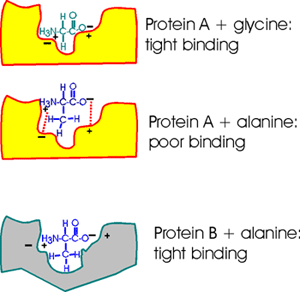
But a similar pocket on the surface of another protein (gray protein B)
could be built to accommodate the methyl (or -CH3 group) of alanine and
supply some more V.D.W. bonds there in the process.
So Protein A binds glycine, but not alanine.
And Protein B binds alanine (but gly not so tightly).
So you can get specific binding ... and this binding is critically dependent
upon the structure of the protein, the shape of the binding site. This
binding is also a critical part of the function of the protein,
and we will soon see.
Specific binding at a protein surface is not restricted to interactions
between a macromolecule and a small molecule. There is also specificity
in the interactions between two macromolecules, as exemplified many times
by quaternary structure: the complementary surfaces of the two correct
subunits fit together with great specificity; just the right subunits
polypeptide specifically associate to form a multimeric protein. For example,
the subunits of immunoglobulin (Ig) never associate with the subunits
of hemoglobin (Hb).
::Domains::
DOMAINS: Now that we have seen that proteins
can bind other molecules with great specificity, I should mention one
last additional aspect of protein structure that I have put off until
now: protein domains. The overall shape of
most proteins is roughly globular, but if one looks more closely, one
can see that most proteins can be divided into sub-regions that are folded
more or less independently of the rest. These folded up globules are called
domains. An interesting feature of many domains is that homologous domains
can often be found in many different proteins. Many of the individual
amino acids in the primary structure are different, but many others are
the same, and the overall shape of the domains in different proteins can
be very similar. A recognizable domain in a protein can often be associated
with a particular function, often the ability to bind a particular small
molecule. In the simple example we used above, a glycine-binding domain
might be found in several different proteins, each of which needs to bind
glycine to carry out its function. Thus we might also have a glucose binding
domain, a phosphate-binding domain or an RNA-binding domain in several
proteins whose function requires them to bind these molecules.
ENZYMES
The ability to bind a specific small molecule is exploited by proteins
when they carry out one their main functions: to act as catalysts that
bring about chemical transformations of the small molecules they bind.
These protein catalysts are called enzymes.
Enzymes represent perhaps the single largest category of proteins, with
respect to function. Since they are responsible for virtually all the
chemical conversions going on in the cell, it is difficult to overestimate
the central role they play in life.
::Catalysis::
Enzymes function as catalysts. So let's define
a catalyst.
Consider the purely chemical reaction between hydrogen gas and iodine
gas:
H2 + I2 --> 2 HI + energy
This reaction goes spontaneously to the right
because H2 and I2 are higher energy compounds than
HI. That is, H2 and I2 are less stable than the combination of these 4
atoms in the form 2HI):
[In the energy diagram below, the ordinate (y-axis) is free energy of
the components, change in free energy (delta G) is only thing that can
be measured, and free energy here is the energy needed to pull apart the
atoms (highest bond strengths will be lowest on the ordinate, as it will
mean more energy has to be put in to raise the atoms to their free, separated,
state)].
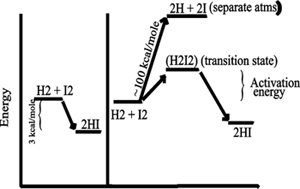
SO if you could invest the energy to separate the atoms, and then let
them fall back to HI's, you would get more energy out (3 kcal/mole difference).
This is a characteristic of a spontaneous
chemical reaction: spontaneous means the reaction can proceed in the direction
indicated (left to right) with the release of energy. In contrast, reactions
that do not release energy, but require energy input, are not spontaneous.
ENERGY RELEASING reactions are called EXERGONIC.
ENERGY-REQUIRING reaction are ENDERGONIC.
{Q&A}
See [Purves6ed
6.5]
Despite the fact that this is an exergonic reaction, it does not proceed
very readily. This failure to react is the case for many such energy releasing
reactions: e.g., burning paper (cellulose + oxygen reaction) can release
much energy, but left to itself in air paper only slowly browns.
We can understand this failure to react if you consider that you'd need
to get the atoms apart before you can rearrange them, and it takes a lot
of energy to break those covalent bonds. Actually, you do NOT need to
take the atoms completely apart:
::Activation
energy::
To get this transformation to proceed, you just need to get to a transition
state. If the two molecules (H2 and I2) collide
at a sufficiently high velocity, then all four of the atoms involved in
the collision can 4 can temporarily form bonds to each other, and this
complex then has a chance to resolve itself into 2 HI (or back in to H2
+ I2):
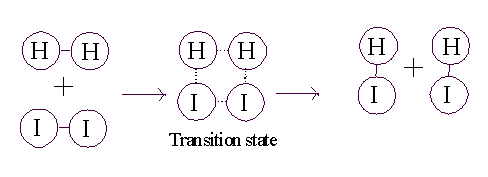
So, for the reaction to proceed, you only need to produce a transition
state, and the energy needed to get to a transition state is called
the ACTIVATION ENERGY.
See [Purves6ed
6.11a], [Purves6ed
6.11b], [Purves6ed
6.11c] and [Purves6ed
6.12].
CATALYSTS ACT BY REDUCING THE
ACTIVATION ENERGY. See [Purves6ed
6.14]. Without a catalyst, you need a forceful collision to get to
the TS. Very few molecules can muster it.
But, for our reaction here, if we add a third substance, if we add some
powdered platinum, the reaction proceeds almost instantly. The platinum
can bind both reactants, so that many of the hydrogen and iodine gas molecules
find themselves as neighbors on the surface of the platinum. More like
bedfellows, as they can be closely packed. So closely, that they can form
a transition state right there on the surface of the platinum particle:
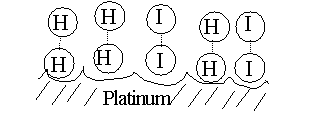
The platinum makes it easier to get to a transition state, no forceful
collision is required, the two participants (reactants) just bind close
together on the common binding surface. And binding to the Pt also weakens
the H-H bond and the I-I bond, making it easier to now form the H-I bond.
The CATALYST IS NOT ALTERED, it just speeds
up the reaction. The catalyst does not change
the situation with respect to the spontaneity of the reaction (energy
releasing character, or DIRECTIONALITY, refer
back to energy diagram), it just speeds things up.
[See animation
- pause lecture audio before you
view this movie - (2 MB file though... probably not worth
the wait unless you are connected to the CU network)].
Chemical catalysts such as Pt can speed things up 10,000 fold, so they
are important in the chemical industry.
(C) Copyright 2001 Lawrence
Chasin and Deborah Mowshowitz Department of Biological Sciences
Columbia University New York, NY
Clickable pictures are from Purves, et. al., Life, 5th Edition,
Sinauer-Freeman's Images of Life 5.0.
A
production of the Columbia
Center for New Media Teaching and Learning