![]() Biology C2005 Lecture 5
Biology C2005 Lecture 5
::Protein 3-dimensional structure::
So the long chain of a polypeptide does not assume
a random coil configuration in space, but, amazingly, folds into a precisely
defined 3-dimensional shape each and every time. That is, each of the
billions of protein molecules of a given kind, with a specific name and
sequence, are superimposable, atom for atom. Well, what is holding the
molecule in this shape? The four weak bond types we discussed earlier,
plus one new bond to be described in a few minutes.
Let's consider how this folding looks in more detail:
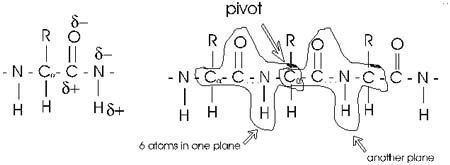
First, the flexible rope was not a good representation of even the backbone,
because the peptide bond itself imposes some constraint on structure.
The peptide bond itself has a property that influences all polypeptides.
Because of the electronegativity difference between C and O and C and
N, there is a partial separation of charge, one you could now have predicted.
What you may not have realized is that the partial + charge on the C and
the partial - charge on the adjacent N, imparts a partial extra bond between
those 2 atoms, and thus a partial double bond character to the C-N bond.
This partial double bond is sufficient to stop free rotation about the
C-N bond (remember the lack of rotation about the double bond in an unsaturated
fatty acid causing a kink?). Thus the backbone is not free to rotate around
all connections, but rather each repeat contains 6 atoms confined to
one plane:
The polypeptide can be visualized as having a series of planes, each able
to rotate about one another. So a chain would be a better representation
than a rope.
::Secondary (2o):
alpha helix::
This partial separation of charge also means that the O and the NH of
the peptide bond can hydrogen bond... to water for example. And since
the N-H is a hydrogen donor and the O is a hydrogen acceptor for a hydrogen
bond, we should consider the possibility that these groups can H-bond
to each other. But H-bonds require a linear orientation of the 3 atoms
involved, so certainly the N-H of the very next residue cannot H-bond
to a C=O preceding it. But what about the N-H on the next residue down
from a given C=O? No, still can't make it. But by the fifth residue down
you are able to line up an NH to the O: -C=O..H-N-. i.e., there are 3
complete residues 3 in between the two residues that are involved in the
the bonding, as shown in the diagram below. (After the
turn between residues 3 and 4, the letters for the atoms have been drawn
backwards, to indicate the that the chain is circling around to achieve
this position. Note that many atoms have been left out, so that the relevant
atoms can be more clearly seen. Note also that the rectangles here delineate
the amino acid residues, not the 6-atom planes discussed above.
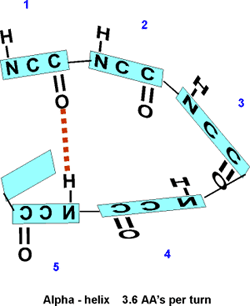
So the C=O of residue #1 can H-bond to the H-N of residue #5. But then
also the C=O of residue #2 should be able to H-bond to the H-N of #6,
and so on. This twisting and H-bonding can hold the backbone in a HELIX,
the so-called alpha-helix.
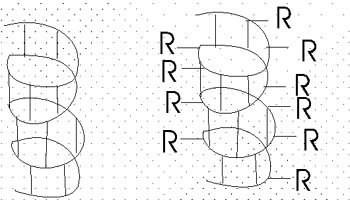
The alpha-helix is an example of secondary structure,
which is (my definition): structure produced by
regular repeated interactions between atoms of the backbone (only).
We might expect all the amino acid backbone
atoms to be in an alpha-helical conformation, but we have left out consideration
of the side chains, which can greatly influence the folding, as we will
see in a minute.
::Secondary (2o):
beta pleated sheet::
The alpha-helix is not the only form of secondary structure, there is
another, the beta-pleated sheet. In this case we once again have the C=O
and the N-H of the backbone forming H-bonds to each other, but in this
case two sections of the polypeptide are aligned side by side:
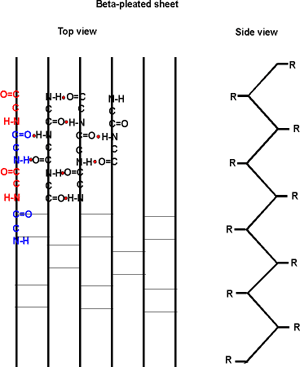
Several sections of polypeptide can line up like this, to produce a sheet
of strands. The chains are usually anti-parallel, but parallel alignments
are also possible. Every other residue H-bonds to a strand alongside;
the side groups stick out both above and below the sheet that is formed
from these H-bonded strands. Once again, side chain interactions play
a major role in allowing or disallowing such secondary structures to form.
But in fact, most proteins do have extensive regions folded into alpha-helices
and beta-pleated sheets.
Secondary structure consists mostly
of these 2 structures, the alpha helix and the beta-pleated sheet.
::Tertiary (3o)=
overall 3-D of a polypeptide::
Tertiary structure means the overall 3-dimensional
folding of a single polypeptide chain. For this overall shape, interactions
between side chains are very important, as are interactions between side
chain and water. A generality is that the
hydrophilic groups are folded to be on the outside where they can interact
with water via H-n bonds, while the hydrophobic side chains are collected
in the inside of the structure, pushed together by hydrophobic forces.
This rule is not at all 100% true, and most proteins have side chains
that deviate from this generality. That is, there are hydrophobic side
chains on the surface, but they are intermingled among the hydrophilic
groups. And there are hydrophilic groups on the inside, where they are
usually interacting with other hydrophilic groups. In fact, it is this
interaction of side chains with each other that confers most of the overall
3-dimensional shape on a given polypeptide.
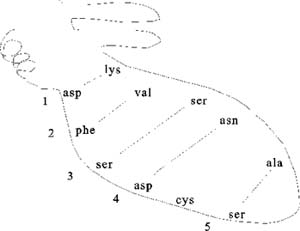
Pictured here are the weak bonds that were introduced earlier. The side
chain interaction indicated in the diagram illustrate examples of these
various interactions. Consult your text for the exact nature of the side
chains:
1. ionic (lys - asp)
2. hydrophobic and VDW (phe - val)
3. H-bond (ser - ser)
4. H-bond to ionic (asp - asn)
5. van der Waals (ser - ala)
Most proteins fold into a roughly globular shape
(enzymes, Hb, antibodies--see a picture
of the enzyme lysozyme: a space-filling model, or showing just the backbone
connections, or a ribbon model), but many take on an elongated or even
fibrous shape (collagen, myosin [in muscle], fibroin [in silk]).
These are weak bonds, but in the aggregate,
they are strong enough to hold the polypeptide together at least under
the thermal motion conditions of physiological temperature (37 deg C).
[Purves6ed
3.8]
::Sulfhydryls, disulfides::
But there is one strong bond that contributes
to the folding of some proteins. This is the DISULFIDE
BOND, and it differs from all these other bonds in being a covalent bond.
It can only be formed between the side chains of two CYSTEINE residues.
The side chain -CH2-SH contains a SULFHYDRYL
group: -SH. Two sulfhydryls can react with oxygen to lose their 2 hydrogen
ATOMS (H with its electron, not H+ ions, not protons) and become bound
to each other in the process:
Protein-CH2-SH + HS-CH2-Protein + üO2
---> Protein-CH2-S-S-CH2-Prot + H2O
So now the two sulfur atoms are sharing electrons in a strong covalent
bond. This bond cannot be broken by mere thermal energy, and so disulfide
bonds hold the two parts of the polypeptide chains that had contained
the two cysteines firmly together. Not all proteins have disulfide bonds,
but many do.
This reaction is an example of an oxidation-reduction reaction: the sufhydryls
are getting oxidized (here oxidation means
losing hydrogen atoms), while the oxygen is getting reduced,
(or gaining hydrogen atoms) and ending up as water. This reaction
will take place rapidly with no further help from catalysts.
(Note that it is not a hydrogen ION (proton, H+) that is being moved about
here, but the hydrogen ATOM with its electron. Actually, it is
the electrons that accompany the hydrogen atoms that are fundamental to
the definition of oxidation/reduction, rather than the hydrogen atoms
as a whole, as we shall see later. That is, oxidation
is the loss of electrons, and reduction
is the gain of electrons, with or without an accompanying
hydrogen ion.)
The net result
is tertiary structure, or the overall 3-dimensional
shape of a folded-up single polypeptide. Note that there will be many
regions of secondary structure within this overall tertiary structure.
It is the interactions of the side chains that are to a large extent responsible
for preventing the whole polypeptide from simply becoming 100% alpha-helix
or 100% beta-sheet.
So now we can see that one polypeptide molecule
can be folded into a compact structure and we can understand what holds
it together, but why is it that there is only one structure formed and
not many? Is there only one solution to the folding problem for a particular
polypeptide chain?
Perhaps all possible conformations are tried in the course of folding,
and only the most stable one accumulates. Can we predict the conformation
from first principles? If we plug in the properties of all the amino acid
side chains, how hydrophobic they are, what is the strength of an ionic
bond, etc. we can ask a computer to try and try many combinations, many
interactions. This is a very difficult computer problem, even for today's
supercomputers, because the number of possibilities for a good-size polypeptide
of say 500 amino acids is enormous (20500). But it has been
tried, and so far usually the wrong structure comes out. The right structure
is determined by examining crystals of the proteins, beaming X-rays through
the protein crystals and calculating how they are refracted by the atoms
in the crystal. Perhaps we really don't know the right properties of the
side chains. Or perhaps there is some guide
to folding that being imposed on the polypeptide as it is being polymerized
in the cell, some outside influence, even a template of sorts, one can
imagine a plaster mold analogy, or some kind of camera lucida.
::Denaturation, renaturation::
Well, if it is true that the folded structure of a particular protein
is unique simply because it is the most stable, then if we UN-fold the
polypeptide, it should be able to RE-fold into its unique structure. How
could we unfold a protein, let's say one with no disulfide bonds, only
weak bonds. We could consider egg albumin, for an everyday case of polypeptide
denaturation. Raw egg white is a concentrated solution of this single
~500 amino acid polypeptide, that exists folded into a roughly spherical
shape.
How can we denature it?....Here's some examples:
Heat: thermal motion becomes to great for the weak bonds.
pH: acids and bases both work, disturbing ionic bonds.
"Chaotropic" agents, such as very high concentrations (e.g., 8M) of urea
(H2N-CO-NH2) can form so many H-bonds that they compete with and disrupt
interactions with water.
organic solvents (e.g., hydrocarbons: octane, benzene, chloroform) turn
the polypeptide inside out, as the hydrophobic forces disappear. {Q&A}
These are all DENATURING conditions. After
denaturing the pure polypeptide, we could try to reverse the disruption.
Let's heat it (boil it). The sphere is now subject to faster and faster
thermal motions, until finally it starts to unravel. What has happened
to the egg white (the albumin polypeptide)? It has become denatured.
[Purves6ed
3.9]. No longer native, which is the structure
in the cell. No covalent bonds have been broken by this 100oC
temperature. The bundled up rope became the open randomly coiled rope
in the Jacuzzi, and this allowed many wrong bonds to form, it exposed
the hydrophobic groups normally hidden in the interior of the protein.
In this concentrated solution a tangled mass of interacting polypeptide
chains was produced, which resulted in a gel, a hot hard-boiled egg. So
while folded up polypeptides are stable enough in their native environment
inside the cell, the 3-dimensional structure is typically rather fragile:
most proteins are easily denatured by heat and other treatments that can
affect these weak bonds. This bundled rope in the Jacuzzi exists on the
verge of becoming unraveled.
So now let's reverse the denaturation, let's cool
down this hard-boiled egg and return it to normal temperatures. The gel
seems to stay. We do not get back our runny egg white. A case of irreversible
denaturation. But not a very fair experiment, letting all those molecules
get SO tangled. Let's try a denaturation - renaturation in a more gentle,
gradual way.
A fellow named Christian Anfinsen did this experiment in the 1950's. He
took a protein called ribonuclease, a protein that is a digestive enzyme,
a protein that helps break down the macromolecule RNA. It must be in its
native structure to do this job.
Actually he had to break disulfides here to get
full denaturation. So he did: he added a reducing agent: mercaptoethanol
(HO-CH3-CH2-SH). In the presence of this reagent, one gets exchange among
the disulfides and the sulfhydryls:
Protein-CH2-S-S-CH2-Protein + 2 HO-CH2CH2-SH
--->
Protein-CH2-SH + HS-CH2-Protein + HO-CH3CH2-S-S-CH2CH2-OH
The protein's disulfide gets reduced (and the S-S bond cleaved), while
the mercaptoethanol gets oxidized.
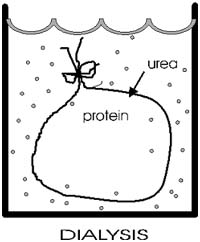
::Dialysis::
After disruption of the disulfide bonds in ribonuclease, Anfinsen placed
the polypeptide in a sack, and added urea, H2N-CO-NH2 to the solution
outside the sack. Urea will break hydrogen bonds at high concentrations
(e.g., 8M). {Q&A}
The sack is made of a semi-permeable plastic material with pores big enough
to allow small molecules like urea and water to pass through but not macromolecules
like albumin or ribonuclease. This process of allowing the concentrations
of changing small molecules to change while holding the concentrations
of large molecules constant is a called dialysis.
After allowing time for diffusion, the concentration of urea inside the
sack should be the same as the concentration outside. {Q&A}
He then checked that the protein had become denatured (e.g., by ultracentrifugation,
see below)..
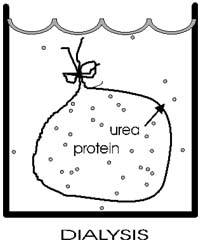
Now he gradually dialyzed out the urea (by changing the solution outside
the sack to stepwise lower and lower concentrations of urea). A dilute
solution of the protein was used, and the gradual removal of the urea
gave time for the polypeptide to re-fold.
He then exposed the polypeptide to oxygen to get back the disulfide from
correctly positioned cysteine side chains (+H20).
He got back native ribonuclease. It checked out physically, and also functionally,
by the fact that it regained its ability to digest RNA.
This type of experiment has been now been repeated
many times for many different proteins. It works for many, fails for some.
But the positive results are very important, for they prove that for many
or even most proteins, all the information that is necessary for the
complex and unique 3-dimensional structure is present in the primary sequence
of the polypeptide chain.
That is, PRIMARY STRUCTURE DETERMINES TERTIARY
STRUCTURE. This conclusion was a major step in biochemistry
and earned Anfinsen a Nobel Prize.
::Chaperonins::
That said, it must be added that in the past 5-10 years, it has become
apparent that some special proteins, called chaperonins,
can help certain other proteins to fold with in the cell. It seems that
these chaperonins may be needed not so much for initial folding, but when
proteins denature inside the cell: for instance, after they have traversed
a membrane, with its hydrophobic environment. Or after cells have been
exposed briefly to slightly elevated temperature (called heat shock),
when a few of the least stable proteins may start to denature. The role,
the generality, and the mechanism by which these proteins aid other proteins
in folding correctly is not yet well-understood. However, these cases
do not really detract from the general principle that primary sequence
CAN determine all higher order structures.
::Quaternary (4o)=
association of multiple polypeptides::
Tertiary structure describes folding of a single polypeptide, and while
many proteins do consist of a single chain, most are composed of several
distinct polypeptide chains. The association of these separate chains
in known as QUATERNARY STRUCTURE.
The number of polypeptides in a protein can be 2, 4, 8 or higher. Or 3
(rarer).
These chains are folded up in 3-dimensions, assuming a tertiary structure,
and then are stuck to each other. What keeps them stuck together? The
same answer as usual: those weak bonds we keep discussing, and more rarely,
the covalent disulfides.
Proteins with quaternary structure are called MULTIMERIC
proteins. Individual polypeptides are called SUB-UNITS
(of the protein).
One polypeptide chain can be considered a monomer, relatively speaking.
A protein with 4 chains a tetramer. Etc. The subunits can be identical
( called HOMOPOLYMERIC) or they can
be different polypeptides (or HETEROPOLYMERIC).
Now we can distinguish a "protein" from a "polypeptide". In its native
form, the macromolecule is called a protein, and may consist of one or
more polypeptides, depending on the protein.
E.g., Hemoglobin, Hb, has the structure a2¤2, consisting
of 4 polypeptides, 2 alpha chains and 2 beta chains, of MW 16,000 each.
So the MW of the Hb protein (a tetramer) is 64,000. [Purves6ed
3.7]
If you denature a multimeric protein, the MW will
change (unless the subunits are held together with disulfide bonds and
you don't disrupt them ), e.g., the MW changes from 64000 to 16000.
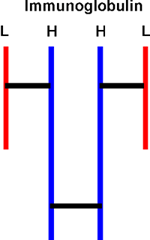
The subunits of some multimeric proteins are held together by disulfide
bonds (in addition to the usual weak bonds). For example, the antibody
molecule, immunoglobulin, is a tetramer of two identical "heavy" chains
(H) and two identical "light" chains (L), or H2L2
and it includes S-S bonds between the H and L chains. You must denature
AND reduce the disulfides to get the individual subunit polypeptides dissociated
from each other.
So the surfaces of polypeptides have also evolved to allow interaction
with other particular subunits but not with other proteins in general.
Consider now Sickle Cell Disease again. Hemoglobin
is a tetramer of 2 pairs of identical sub-units: a2b2.
Glu --> val was the a.a. change comparing normal Hb to sickle Hb (HbS).
The result is that the tetramers inappropriately interact, presumably
via hydrophobic interactions that in normal Hb is precluded by the charged
glu. In HbS this position is valine and now has a more hydrophobic patch
of surface. The result is that these patches can now get stuck together
by hydrophobic forces, and aided by the fact that each HbS molecule has
two such patches (one for each beta chain), and the concentration of Hb
molecules inside a red blood cell is very high (they can almost be viewed
as bags as Hb). You get long chains of tetramers, and these long arrays
can distort the shape RBC (red blood cell) into a sickle shape. This shape
is not a hydrodynamic as the original c shape, and the RBCs can now get
clog in small capillaries, the manifestation of the disease. One a.a.
out of 250 was responsible. Once again we see that proteins are fragile,
are often only on the brink of stability.
PROSTHETIC GROUPS: There are some NON-amino
acid components of proteins that are so tightly bound they are considered
part of the protein. These small molecules are usually essential for the
function of the protein. For example, in hemoglobin, the "heme" groups
are actually organic ring compounds with an iron atom at their center,
and it is this iron atom that actually binds the oxygen that is carried
by the hemoglobin protein. Some of the vitamins become prosthetic groups
(e.g. riboflavin). See B: 427 for heme structure.
PROTEIN
PURIFICATION (SEPARATIONS)
::Protein purification methods: Ultracentrifugation::
While we are on the subject of proteins, let's take some time once again
to discuss methodology. In this case, the purification of individual proteins,
which involves their separation from all the other proteins in the cell.
Much of what we want to know about proteins requires that we have a pure
preparation contain only protein molecules of one homogeneous type. Since
there are 3000 different types of protein molecules in E. coli, our task
will be to separate one away from all 2999 others, to purify it.
[The word separate sometimes causes confusion at this point. In the context
of purifications, "separate" is used as a relatively passive action, operating
on a mixture without altering the components greatly, e.g., to separate
the wheat from the chaff. Our primary objective here will not be to cleave
molecules ("I'm gonna separate your head from your body"), although some
cleavages may occur in he course of an experiment (e.g., cleavage of the
disulfides of immunoglobulin in order to effect a separation of the individual
subunits).]
How can we proceed to purify a protein? Well, what makes one protein different
from another?
Can you proffer some characteristics?: size (MW), charge (net),
shape, hydrophobicity (solubility), surface binding ability....
Yes, all these are used in what is still a challenging task for any biochemistry
laboratory, the purification of its favorite protein.
Here is one sometimes useful method: ULTRACENTRIFUGATION
ultra means = >20,000 rpm; 60,000 rpm is common, compare. a Ferrari at
6000 rpm, redlining; this is ten times faster; you need a vacuum chamber
so no heat from air friction can be produced. )
Diagram of tube, spin, distribution of molecules ... 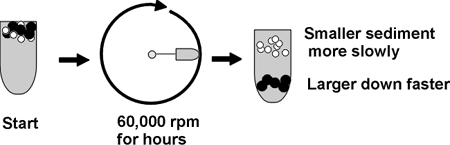
A mixture of molecules will be subject to two main forces in the ultracentifuge
as it starts to spin (ignoring buoyant force):
Causing sedimentation is the centrifugal force
= m(omega)2r = (which is proportional to the mass or MW of a protein).
m = mass, omega = angular velocity, and r = distance
from the center of rotation.
Opposing sedimentation = friction = foV.
fo = frictional coefficient, a constant
for any particular protein, it is minimum for a sphere, higher for less
compact shapes like cigars or pancakes. V = velocity
of the molecule as it moves away from the center of rotation.
Soon after accelerating, V increases to a point where no further acceleration
takes place, as the forces on the molecule are balanced. It continues
to sediment, but at a constant velocity.
Now at this point, at this velocity: Centrifugal Force = Frictional force
(there's no net force, no acceleration, but
constant velocity)
So at this point (soon achieved): M(omega)2r
= foV
And: V = m(omega)2r/fo,
where f = a frictional coefficient dependent on shape (to visualize the
effect of shape on friction, compare the velocity of a falling feather
vs. a tiny pebble of equal weight, dropped in the fluid of air).
Higher f = more friction.
If we assume a spherical shape, then we can estimate a MW (Assume fo,
and then measure V and r, so we can solve for m, or the MW)
On the other hand, if we know the MW, we can get information about shape
(via fo).
Sedimentation velocity is often measured in Svedbergs, which takes the
centrifugation conditions into account s = V/(omega)2r, and
so m = sfo.
So ultracentrifugation separates proteins on the
basis of MW and shape. It is a gentle procedure (non-denaturing, can be
carried out at nice low temperature (say 4 deg C, which tends to stabilize
proteins) and in the presence of a buffer at pH 7 and physiological levels
of salts).
You can recover your protein by punching a hole in the bottom of the centrifuge
tube, and collecting the solution in a series of tubes as it drips out
the bottom. Each tube can then be examined, or assayed, for the presence
of the protein to be purified. For this purpose you need to be able to
detect the protein in the midst of the other proteins. For example, if
you were purifying Anfinsen's ribonuclease, you could measure the ability
of the tube contents to catalyze the breakdown of RNA to its monomers.
How about separation on the basis of the net charge of a protein. We separated
amino acids on the basis of charge in paper electrophoresis. For proteins,
the solid supporting material is a gel, not paper:
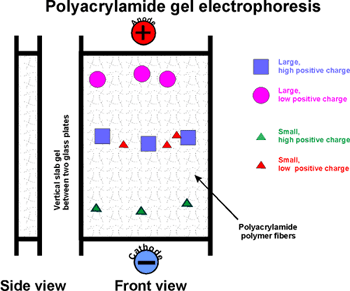
GEL ELECTROPHORESIS:
There are two types -
::Native gel electrophoresis::
First: native gel electrophoresis.
Acrylamide (a monomer in this chemistry) in aqueous solution ---> polyacrylamide
(P.A.G.E.). The result is a network of polymer
fibers, which form a gel, with the consistency ~ Jello.
Usually a vertical apparatus, with an anode and a cathode. Apply the protein
mixture to the top of a slab of this gel.
Apply voltage (~200 v).
The gel consists of a tight fiber network, so proteins have trouble migrating,
negotiating their way through the tangled fibers.
Their rate of migration depends on two properties:
Their net charge and their "size" (which is
proportional to MW if spherical)
Molecules with the most charge (net) (of a sign opposite to that of the
far electrode) migrate to the far electrode fastest.
Molecules that are smallest (i.e., lowest MW) can worm their way through
the gel fibers fastest. So the smallest and most highly charged wins the
race.
After the electrophoresis has been stopped, molecules will be distributed
along the gel length according to these two characteristics (MW and net
charge).
[Note that molecules with a charge opposite to the near electrode, will
migrate up and off the gel, into the buffer reservoir and be lost. Trial
and error will dictate how you setup the electrophoresis if you do not
know the charge on the protein you are trying to isolate.]
::SDS gel electrophoresis::
Second, a more widely used variation of gel electrophoresis: SDS
PAGE.
Add sodium dodecyl sulfate, SDS (or SLS): CH3-(CH2)11-
SO4=
[sulfate is similar in structure to phosphate, and is a strong acid].
Like a phospholipid, SDS has a highly polar end and a highly hydrophobic
body.
Might you expect SDS to denature a protein? Yes. It's a detergent and
a powerful denaturant. It binds all over the protein, coating every protein
with a uniform negative charge. SDS is put
into in the gel when you form it and into the electrophoresis buffer.
Now run SDS-PAGE. Where should the anode be placed? Does it matter? Yes,
the protein is coated with negative charge now so anode is always at the
bottom.
Under these denaturing conditions, the polypeptides exist as a random
coils, which then migrates solely on the basis of their size, which is
the equivalent of a sphere for all polypeptides. Larger molecules have
more difficulty finding their way through the polyacrylamide fibers. So
the lowest MW wins.
One must remember to reduce the disulfides with mercaptoethanol first
(usually), so as to have a truly random coil to compare.
If you run standards of known MW, you can determine the MW of your protein
by comparison, and this is a very common way to assign a MW to a polypeptide.
However, it is not always completely accurate, as some proteins probably
do bind a bit more SDS than others.
If you don't yet know what a protein does, you can just call it by its
molecular weight, from SDS gels: e.g., p53, a famous protein whose absence
is associated with cancer was named this was, and the name has stuck even
though quite alot is known about its function (p in p53 stands for protein,
so you have names like p27, p100 etc.).
::Gel filtration::
If we want to know the MW of a protein in its native, even quaternary
structure?
For this we could use molecular sieve chromatography,
or Sephadex, or gel filtration
(these are all ~synonymous).
You start with plastic beads in a glass column with a support screen on
the bottom.
Add your protein mixture to the top. Elute with a buffer. The beads are
riddled with channels of a specified size. If a protein is smaller than
the channel size, it enters, explores, diffuses out finally, having wasted
its time in the race to the bottom of the column. Larger proteins can't
fit in to the channels, don't waste their time, and win the race. Intermediate
sizes waste some time but less than the smaller proteins. So larger molecules
come out (elute) first, and the smallest come out last. Here again, you
would collect the eluted proteins in a series of tubes, and then assay
each tube for the presence of the protein being purified. If you calibrate
the column by noting the behavior of spherical proteins of known size,
you can determine the MW of your protein by comparison, if it is also
spherical. If is is not spherical it will appear to have a higher molecular
weight than its true MW (imagine a pancake being excluded from a channel
while a sphere of the same MW gets in).
Other methods include ion exchange chromatography, which also takes advantage
if the net charge on a protein, and affinity chromatography, which takes
advantage of the surface properties of a protein (which we'll discuss
next). One can purify a particular protein away from all other proteins
in 4-5 such steps. For more on protein separation techniques, see the
protein separation handout.
(C) Copyright 2001 Lawrence Chasin and Deborah
Mowshowitz Department of Biological Sciences Columbia University
New York, NY
Clickable pictures are from Purves, et. al., Life, 5th Edition,
Sinauer-Freeman's Images of Life 5.0.
A production of the Columbia
Center for New Media Teaching and Learning